Fda Guidance Medical Device Reporting
FDA Finalizes Guidance November 9 2016 Under the Medical Device Reporting MDR regulation there is a mechanism that allows FDA and device manufacturers to identify and monitor adverse events deaths and serious injuries and certain malfunctions of devices to detect and correct problems in a timely manner. The Agency also considers the history of adverse event reports under the medical device reporting program for these devices as well as their history of product recalls.
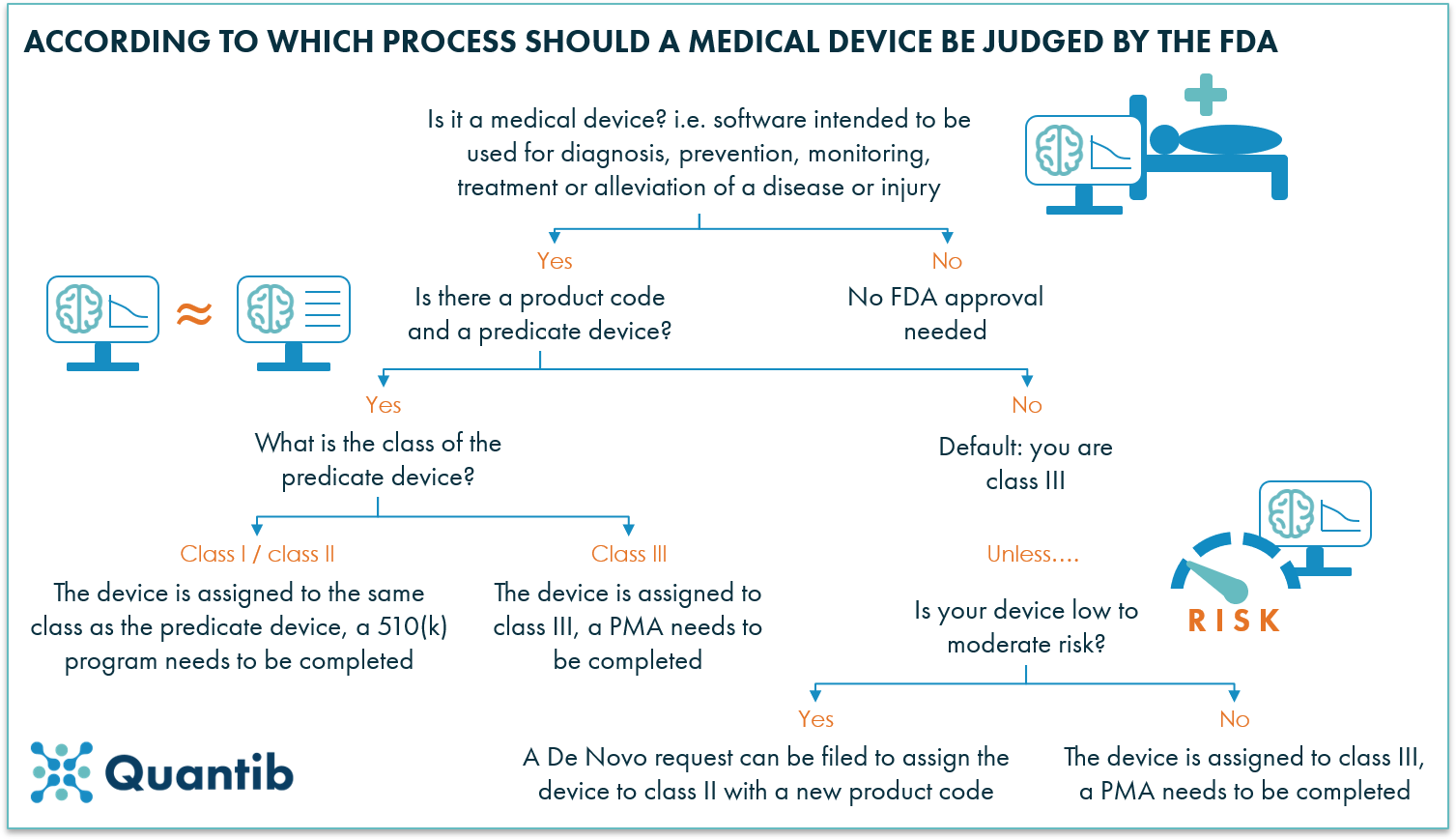
A 101 Guide To The Fda Regulatory Process For Ai Radiology Software
Manufacturers - Medical device manufacturers are required to file a report with the FDA whenever they learn that one of their devices may have caused or contributed to a death or serious injury.
Fda guidance medical device reporting. FDA Gives Guidance on Reporting Medical Device Shortages Posted on May 14 2020 by Rebecca Trela Last week FDA released guidance for life sciences manufacturers that produce medical devices and components critical to public health including materials that support or sustain life or are used in emergency care or surgery. Final Rule on Electronic Medical Device Reporting eMDR Electronic Medical Device Reporting eMDR On Feb. This document supersedes Medical Device Reporting for Manufacturers dated March 1997.
13 2014 the FDA published a final rule on Electronic Medical Device Reporting. A reportable death serious injury or malfunction is based on information a manufacturer receives or otherwise becomes aware of from any source which reasonably suggests that one of its marketed devices. Determination Regarding Surgeons Gloves and Patient Examination Gloves and Premarket Notification.
FDA Notification and Medical Device Reporting for Laboratory Developed Tests LDTs DRAFT GUIDANCE. These reports have generally been required for the first three years that a new device is on market in Taiwan. Based on the risks inherent with surgeons gloves and patient examination gloves and the diseases being prevented FDA.
Manufacturers of medical devices subject to the reporting requirement should notify FDA of a discontinuation or interruption within seven calendar days of the disruption occurring. This guidance represents the current thinking of the Food and Drug Administration FDA or. Manufacturers of medical devices are required to submit reports to FDA of a reportable death serious injury or device malfunction.
This requirement no longer exists. Below we describe the FDA medical device reporting requirements as they apply to each group. FDA released a guidance on medical device reporting MDR an important document with which manufacturers should familiarize themselves.
The FDA encourages health care personnel to report any adverse events or suspected adverse events experienced with any medical devices including decontamination systems bioburden reduction. The Medical Device Reporting MDR regulation 21 CFR Part 803 contains mandatory requirements for manufacturers importers and device user. Manufacturers must also report device malfunctions when they believe that a death or serious injury could occur if the.
Mandatory Medical Device Reporting Requirements. US FDA finalizes post-market safety reporting guidance for combination products Aug 1 2019 Final guidance issued by the US Food and Drug Administration explains how combination product manufacturers should comply with post-market safety reporting requirements originally established in. Draft guidances detail medical device vigilance reporting procedures.
Draft Guidance for Industry Food and Drug Administration Staff and Clinical Laboratories CDRHCBER October. FDA Notification and Medical Device Reporting for Laboratory Developed Tests LDTs. The deadline stems from FDAs interpretation of the CARES Act requirement for companies to send a notification as soon as is practicable A manufacturer should only consider internal capacity not industry-wide.
Medical Device Reporting for Manufacturers. This document provides general guidance regarding the reporting of adverse events required by the Medical Device Reporting MDR Regulation. The first guidance Medical Device Safety Surveillance Management Measures links in Chinese defines the requirements surrounding submission to the TFDA of Periodic Safety Update Reports PSURs.
This guidance document is being distributed for comment purposes only. It outlines expectations of reporting and it is a vital part of post-market surveillance.
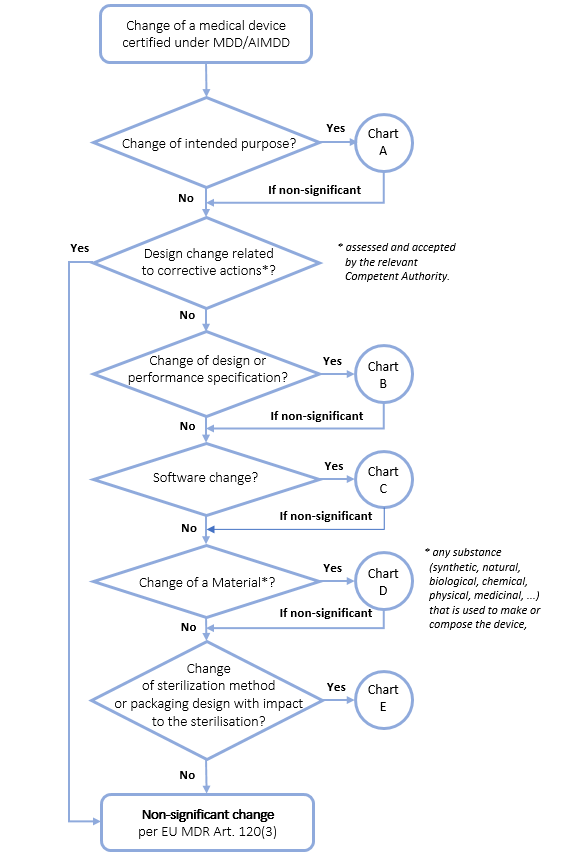
Mdr Guidance On Significant Changes For Medical Devices
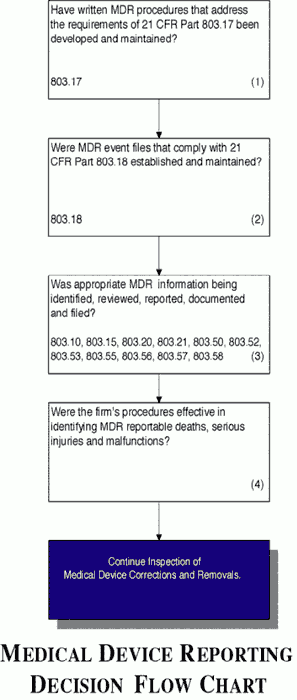
Medical Device Reporting Fda

Complaint Management And Medical Device Reporting Overview
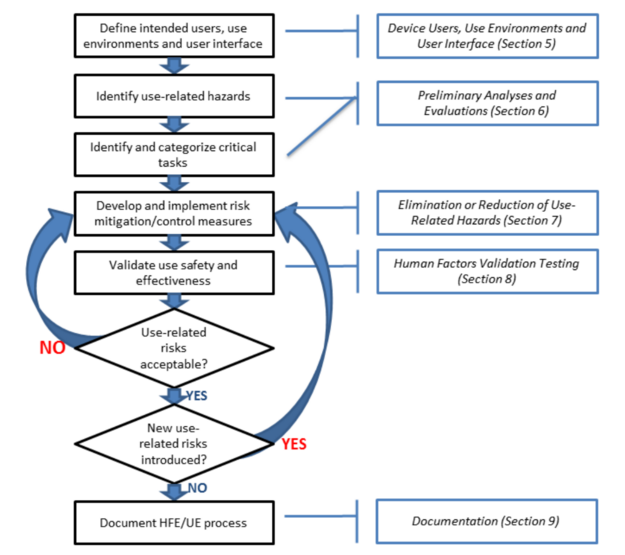
Usability Engineering And Human Factors Testing For Devices Medical Device Academy
{kind=link}