Fda Medical Device Adverse Event Reporting
If an adverse event occurs in the United States Emergo can assist you in submitting an Electronic Medical Device Report eMDR to the FDA using their web-based Electronic Submissions Gateway ESG. An adverse event can refer to any unfavorable occurrence involving your medical device in.
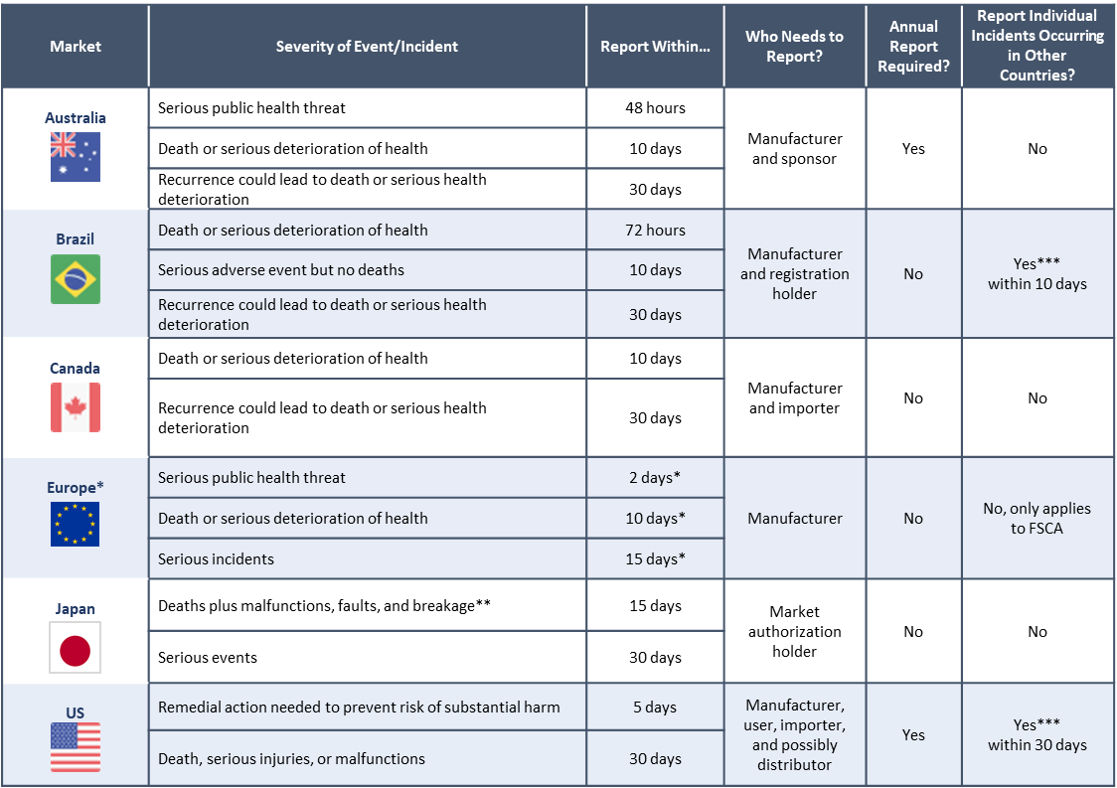
Medical Device Incident Reporting Timelines In 6 Major Markets
FDA Adverse Event Report Form You can obtain the FDA Adverse Event Report Form MEDWATCH form FDA 3500A from the FDAs MedWatch Adverse Event Reporting program on their web site httpswwwaccessdatafdagovscriptsmedwatch.

Fda medical device adverse event reporting. As is the case for conventionally FDA-cleared or approved devices adverse event reports for EUA-designated devices should be submitted electronically and in accordance with the agencys guidance on electronic medical device reporting. MDSAP Medical Device Adverse Events and Advisory Notice. FDA Releases 20 Years of Data on Medical Device Adverse Event Reports Posted 21 June 2019 By Zachary Brennan The US Food and Drug Administration FDA recently ended its Alternative Summary Reporting ASR program for medical devices revoked the related exemptions and on Friday made available on its website all adverse event reports received under ASR exemptions from 1999 to 2019.
The data consists of voluntary reports. The form can be completed online or printed out. Final FDA rules clarify adverse event reporting for contract manufacturers November 14 2016 By Brad Perriello The FDA last week issued final guidance for medical device companies on the requirements for reporting adverse events that walked back much of the burden for contract manufacturers.
If an adverse event meets the criteria for reporting the ASF must report that event regardless of the nature or location of the medical service provided by. Manufacturer and User Facility Device Experience MAUDE data MAUDE data contain reports received by the FDA of adverse events involving medical devices. This is a clip from a recent Q1 Productions Webinar on Medical Device Reporting of Adverse Events to the FDA.
Adverse Event Reporting for EUA Medical Devices Jun 16 2020 The Food and Drug Administration FDA or the Agency issued detailed guidelines dedicated to adverse event reporting rules for medical devices placed on the market under the Emergency Use Authorization EUA. The openFDA device adverse event API returns data from Manufacturer and User Facility Device Experience MAUDE an FDA dataset that contains medical device adverse event reports submitted by. The Medical Device Reporting MDR regulation 21 CFR Part 803 contains mandatory requirements for manufacturers importers and device user.
To report problems with animal drugs devices or foods including pet foods Report a Problem to the Center for Veterinary Medicine. Each year the FDA receives several hundred thousand medical device reports MDRs of suspected device-associated deaths serious injuries and malfunctions. Under the Medical Device Reporting MDR regulation there is a mechanism that allows FDA and device manufacturers to identify and monitor adverse events deaths and serious injuries and certain malfunctions of devices to detect and correct problems in a timely manner.
Animal Drug Device and Food Product Problems. Mandatory Medical Device Reporting Requirements. A safety report or other information submitted by a sponsor under this part and any release by FDA of that report or information does not necessarily reflect a conclusion by the sponsor or FDA.
The webcast covered How to properly report. The FDA uses MDRs to.
Complaint Management And Medical Device Reporting Overview

Medical Device Reporting Mdr Electronic Mdr Emdr Reporting Process Usfda

Medical Device Reporting Mdr Electronic Mdr Emdr Reporting Process Usfda
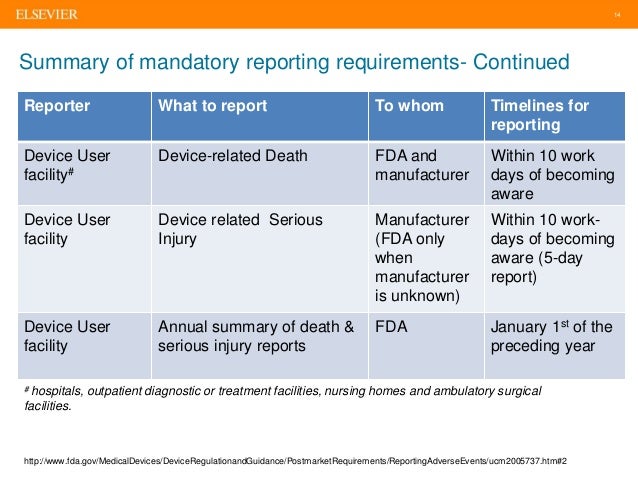
Medical Device Reporting 27 Sep2016
{kind=link}